

腸内細菌のデータは、しばしば高次元、異なる構造(カウントや割合)、明らかな非正規分布、過分散および過剰なゼロの存在…などといった特徴を持っており、特別な解析手法が必要な場合が多い。そこで、活用できるパッケージの一つにMaAsLin2というとても便利なパッケージがある。しかし、オプションが非常に多く、それらを適切に選択しなければ誤った結論を導いてしまうかもしれない。そこで、ここに簡単な使用方法、オプションを選択する際のポイントなどをまとめることにした。
MaAsLin2とは?
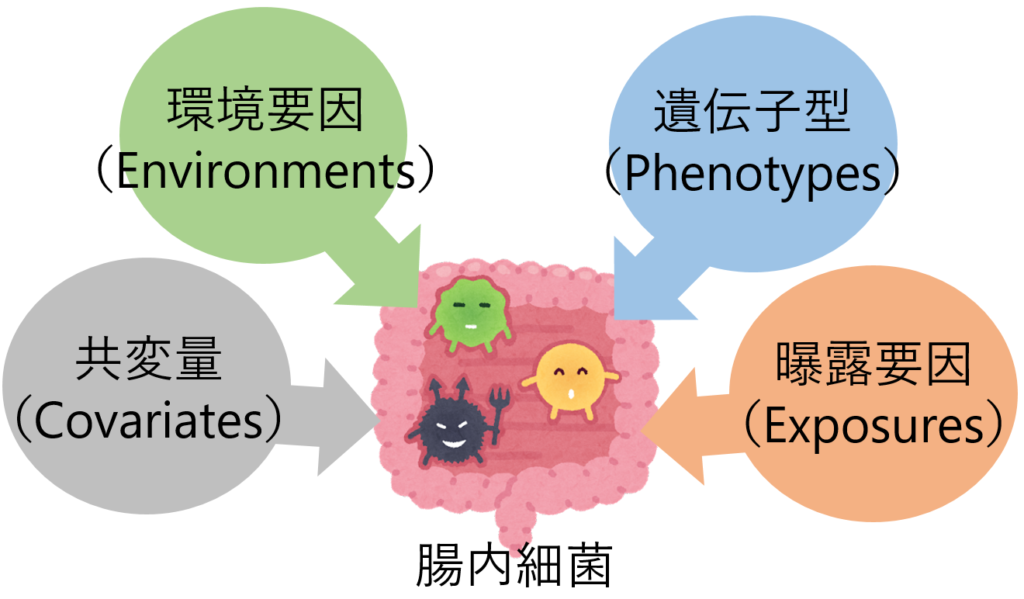
MaAsLin2( Microbiome Multivariable Association with Linear Models ver.2)は、環境要因、遺伝子、曝露要因、共変量と腸内細菌叢の特徴など、多変数間の関連を明らかにするための包括的なパッケージである。また、統計解析と同時にグラフ化が可能で、横断的または縦断的データの両方の解析に使用できる。
シミュレーション研究において、MaAsLin2がデータの特徴をとらえ、偽発見率(FDR)は抑えつつも、検出力(パワー)は維持できたことが報告されている¹。
サポートが充実
MaAsLinは、独自のフォーラムやGoogleフォーラムを用意しており、不明な点があれば直接専門家からの解答を得ることができる。質問と回答をざっと見るだけでも、より実践的な使用方法を学ぶことができる。
インストール方法
以下のコードでインストールできる。
if(!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("Maaslin2")※インストールできなかった場合
インストールできない場合は、RまたはBioconductorパッケージのバージョンが古い可能性があり、それらをアップデートすることにより解決する場合がある。ちなみにそれぞれのバージョンRは3.6.0.以上、Bioconductorは3.10以上である必要がある。
Bioconductorパッケージのアップデートは以下のコードにより実行できる。
BiocManager::install(version = "3.10")なお、インストール以降のコードの例はこちらのページ(外部リンク)を参照してください。
解析に必要なデータファイル
MaAsLinを用いた解析では、主に二種類のデータファイルが必要である。
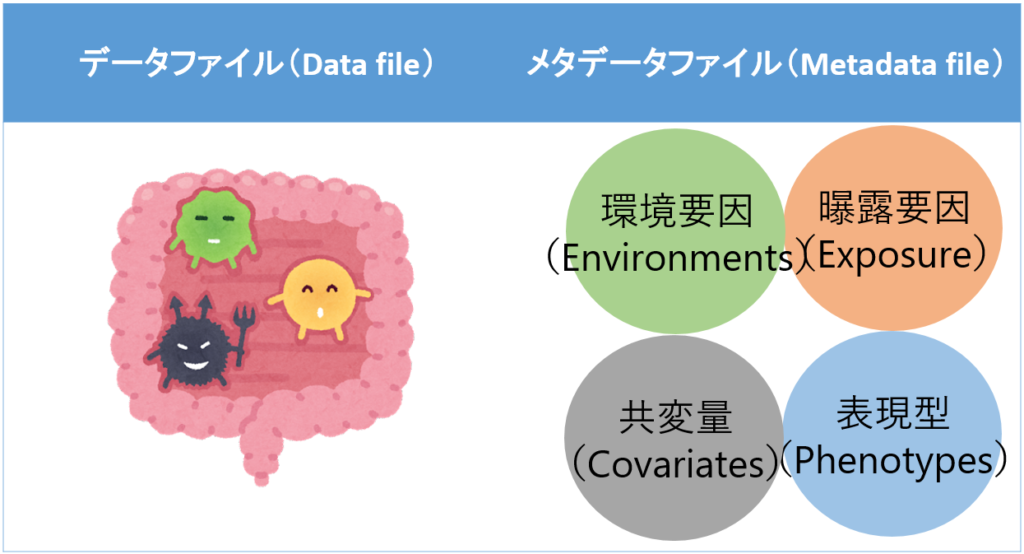
| データファイル | 分類学的または機能的な腸内細菌の豊富さを示すデータを含む(カウントデータ、割合データなど) |
| メタデータファイル | 環境要因、曝露要因、共変量などのデータを含む(介入の有無、居住地域、性別、年齢、食習慣など) |
- それぞれのデータファイルは、行が対象者を示す変数(IDなど)、列はデータファイルであれば腸内細菌の属名や種名、機能、メタデータファイルであれば関心のある変数とする。
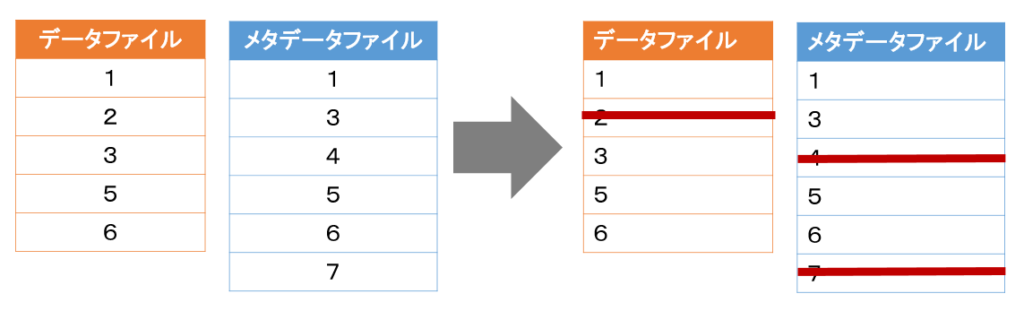
- 上記の二つのデータファイルに含まれるサンプルの数は異なっていても良い。なお、両方のデータファイルに存在しないサンプルは自動的に削除される。
MaAsLinで使用できるモデルとオプション
MaAsLinでは、様々なモデルを用いて解析を行うことができる。デフォルトでは最も適した手法が選択されるようだが、必要に応じて変更することが可能である。なお、データファイルに入力されているデータの種類によって、使用できる/使用すべきモデルが異なる。
モデル一覧
| NEGBIN (Negative binomial) | ※カウントデータのみ使用可(割合、相対的存在量のデータなどは不可) |
| ZINB (Zero-Inflated Negative Binomial) | ※カウントデータのみ使用可(割合、相対的存在量のデータなどは不可) |
| LM (Linear Models) | このモデルのみ負の数、正の数どちらも使用できる。 |
| CPLM (Compound Poisson Linear Models ) | 正の数のみ使用できる。 ゼロが過剰な場合により良いパフォーマンスを発揮する可能性があるが、まだLMより優れているかについて十分な検証はされてない¹。 |
オプション
モデルに加えて、データの正規化や変換方法をオプションとして選択することができる。なお、どちらの場合も特に指定しない場合は”NONE”とする。指定しないと、おそらくデフォルトの設定が自動的に設定される。
正規化方法の一覧(Normalization methods)
TSSは、最もシンプルな計算方法で、サンプルごとのライブラリサイズ(リード数)の違いは考慮しない。その一方でTSSやCSS、CLRではサンプルごとのライブラリサイズを考慮した正規化の方法である。
| TMM (Trimmed Mean of M-values) | |
| TSS (Total Sum Scaling) | 各サンプルの存在量を合計でスケーリングし、相対的な存在量に変換する。 デフォルトではこれが適用される。シミュレーション研究によれば、TMMが他の方法と比べて最もGoodな方法だった¹。 |
| CSS (Cumulative Sum Scaling) | サンプルごとのライブラリサイズの違いを考慮する |
| CLR (centered log ratio) | サンプルごとのライブラリサイズの違いを考慮する |
| NONE (正規化しない) | 正規化しない場合や、すでに正規化されているデータを用いる場合はこれを選択する。 |
変換方法の一覧(Transformation methods)
| LOG (Log transformation) | 対数変換 |
| LOGIT (Logit transformation) | 確率のオッズの対数 |
| AST (Arcsine Square Root) | アークサイン変換 二項分布のような変数を正規分布に近づける数値変換法²。 ノンパラメトリック検定で良く用いられる。 |
| NONE (変換しない) |
実用例
PERMANOVAと組み合わせて使う
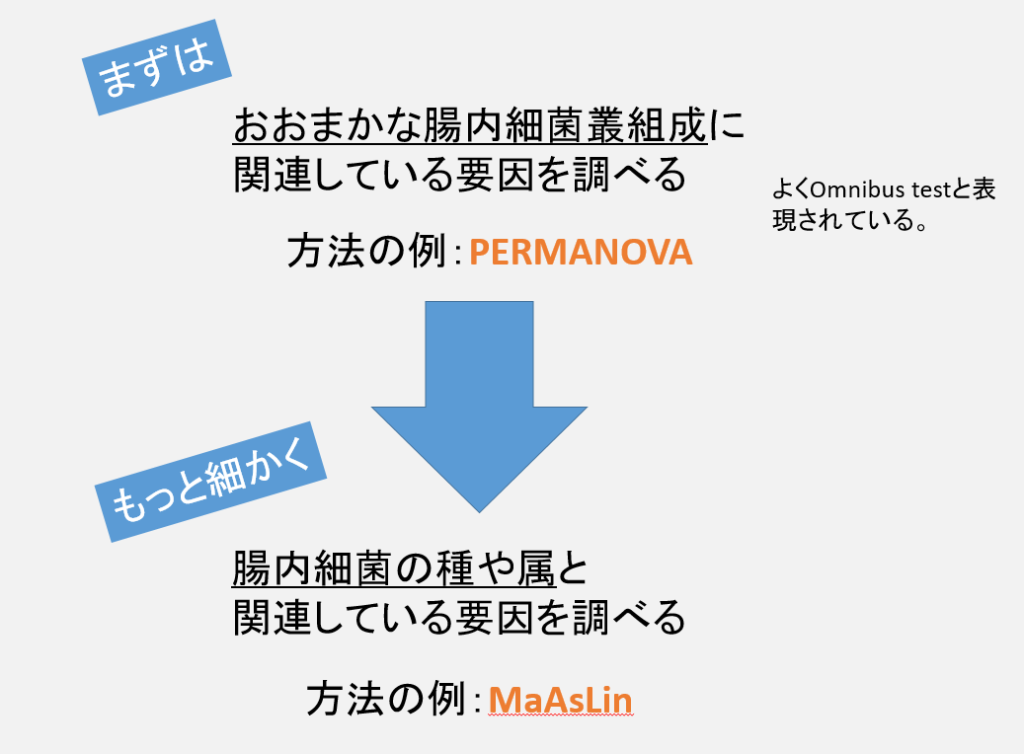
論文を読んでいると、PERMANOVAを使っておおまかな腸内細菌叢に関連する要因を探し出し、その次にMaAsLinを使ってより詳しい関連(属や種)を調べるという方法をとっているものがいくつかあった。
たとえば、喫煙が腸内細菌叢に関連することがわかったとする。そうすると次に疑問となるのは、喫煙が腸内細菌叢を構成するどの菌に関連しているのか?である。例えば、この論文では腸内細菌叢にするCRPや食物繊維摂取の関連を解析している。
Li X, Kimita W, Cho J, Ko J, Bharmal SH, Petrov MS. Dietary Fibre Intake in Type 2 and New-Onset Prediabetes/Diabetes after Acute Pancreatitis: A Nested Cross-Sectional Study. Nutrients. 2021 Mar 29;13(4):1112. doi: 10.3390/nu13041112. PMID: 33805259; PMCID: PMC8066410.
https://pubmed.ncbi.nlm.nih.gov/33805259/
References
- Himel Mallick, Ali Rahnavard, Lauren J. McIver, Siyuan Ma, Yancong Zhang, Long H. Nguyen, Timothy L. Tickle, George Weingart, Boyu Ren, Emma Schwager, Suvo Catterjee, Kelsey N. Thompson, Jeremy E. Wilkinson, Ayshwarya Subramanian, Yiren Lu, Levi Waldron, Joseph N. Paulson, Eric A. Franzosa, Hector Corrada Bravo, Curtis Huttenhower. “Multivariable Association in Population-scale Meta-omics Studies”. BioRxiv Preprint: https://doi.org/10.1101/2021.01.20.427420
- Ⅵ.統計処理. http://www.nlbc.go.jp/gijutumanyuaru/manual16/16-7.pdf. 2021.01/11 閲覧.